1.はじめに
細胞内に発現しているDNA/RNAを可視的に検出する手法としてin situ hybridization法が開発され、普及してきている。In situ hybridization法は、はじめに放射性リンで標識したcDNAプローブを用いる手法(hot法)が開発された。しかしこの古典的手法は、DNAプローブを用いるために検出の特異性が低く、放射性リンで標識するために細胞レベルで高い局在性を保ちながら高感度に検出することが困難であった。Digoxigenin(DIG)で核酸を標識する手法(cold法)が、Roche Diagnostics社(Rotkreuz、 Switzerland; Boehringer Mannheim社が開発してきたものだが、最近Roche社に統合された)を中心に開発され、これを用いてcRNAプローブを標識することで特異性高く、かつ局在性を保ちながら非常に高感度に検出できるようになってきている。しかしながら、高感度なDIG法を用いても、数コピーのレベルの極僅かしか発現していない核酸をin situ hybridization法で検出することはできない。そこで、あらかじめ標的とするDNA(RNAを検出しようとする場合は、あらかじめ逆転写してcDNAにしておかなくてはならない)をpolymerase chain reaction(PCR)法で数百万倍にまで増やしておき、その後in situ hybridization法で検出するin situ PCR法が開発された。In situ PCR法で組織切片に含まれる標的DNA/RNAを可視化しようとする場合、0.1 pg/10,000μm2(厚さ3μmの切片の場合)が検出限界であると考えられている。In situ PCR法は、検出しようとする標的核酸の増幅法の違いから、大きく2種類の手法に分けられる。
(A-a)ひとつは、特異的なプライマーを設計し、標的DNAだけを特異的に増幅しておき、これをin situ hybridization法で検出する手法である。この手法は特異性が高くて理想的であるが、in situで組織構造を破壊することなく標的DNAだけを特異的に増幅する条件を決定することが容易でないので、宿主遺伝子に潜り込んだ数コピーのウイルスを検出するような高い特異性が求められる場合に用いられている。今回は紙面の都合で触れない。
(A-b)A-a法の変法として、組織切片上にある標的DNAをPCR法にて増幅する過程で、増幅されるDNAに直接DIGなどで標識したヌクレオチドを組み込むことで、増幅後のISHの過程を省略して標的DNAを可視化する手法が開発されている。この手法は、A-a法と比較してプロセスが少ないので組織構造の保存性が高く、かつ非特異的反応が少ない点が優れておりウイルスの高感度検出法として優れている。初めてin situ PCRを試みる場合には適した手法であるので、今回はこの手技を説明する。しかしながら、PCRで増幅される分子の特異性が保証されなくてはならないので、細胞内に発現している多くのmRNAを検出しようとする場合には、特異的なプライマーの設計や標的DNAだけを特異的に増幅できるPCR条件の決定などの実験条件の設定が容易でないことを充分に理解し、常に検出されたものの特異性を確認するように細心の配慮をすることが重要である。
(B)もうひとつは、7塩基ほどのランダムプライマーを用いて標的核酸を含む全てのDNAをあらかじめ増幅しておき、その後in situ hybridization法で標的核酸を検出する手法である。前者と比較して後者は比較的容易な手法で、非常に発現量が少ないmRNAを検出しようとする場合には優れているので繁用されるようになってきているが、ウイルスの検出のように高い特異性を求められる場合には多くの問題点を含んでいる。今回は紙面の都合で触れない。
一般には前者(A-aとA-b)と後者(B)をあわせてin situ PCR法と呼ぶが、両者を区別するために前者をin situ PCR法、後者をPCR-in situ hybridization(PCR-ISH)法と呼びわける場合がある。歴史的には、最初にPCR-ISH法が開発され、続いてPCR-ISH法の変法としてin situ PCR法が開発され、その変法としてPCR後のin situ hybridizationが省略できるPCR過程で標識ヌクレオチドを組み込む手法が開発された。
In situ PCR法は、組織切片や細胞標本などに適用できるが、本稿では組織切片を対象に述べる。In situ PCR法は、生化学的プロセス(プライマーの設計・作製と標識)と組織・細胞化学的プロセス(組織切片の準備、切片上でのPCR反応の実施、反応産物の可視化反応)とからなる。前者は一般のPCR条件の決定と同じであるので、紙面の都合上省略する。
2.組織切片の調製
組織切片でin situ PCRを行う場合のヌクレオチド標識は発色反応を顕微鏡下に精密に制御できるDIG標識法を採用することを推奨する。Roche Diagnostics社からkitが販売されている。高価であるが簡便に実験できるのでこれを使用するとよい。Hotラベル法は、感度、局在性保持ともにDIG法におよばず、時代遅れである。
■チップ:Roche Diagnostics社から日本語で書かれた懇切丁寧なDIG標識法のマニュアル冊子が提供されている。これはいかなる市販の実験書より優れている。これの盗作としか思えないような日本語の実験書が最近出版されている。In situ PCRを始める方は、最初に試薬代理店を通じて入手し、熟読した後実験されるべきである。この点がこのテクニカルチップスの唯一役立つポイントと思う。
(2-1)組織切片の作製
言うまでもないことだが、mRNAの検出を目的とする場合には、RNA分解酵素の活性を完全に阻害しなくてはならない。以下の各操作は、可能ならRNA分解酵素で汚染していない専用実験室で専用の試薬、器具、容器を準備して行うことが望ましい。使用する水は、特に記述していない場合は0.1%
(w/v) diethylpyrocarbonate (DEPC)-ミリQ水を用いる。
■チップ:最も簡便なRNA分解酵素の阻害法として、台所用の塩素系漂白剤を蒸留水にて10倍希釈し、これにミクロトーム替刃などなどの小さな器具を浸漬した後実験に供する。ミクロトームなどの大型器具は希釈液を含ませた清浄なガーゼで丁寧に拭く。清浄し易いように厚板ガラスの上にミクロトームなどの薄切用の機器一式をセットして使用するとよい。なお、塩素は有毒なので、実験室の換気をこころがけること。
■チップ:プロセスの多い組織化学的染色法の宿命であるが、in situ PCR法も偽反応が出やすい。常に1枚の同一スライドグラス上に観察用、positive
control用、negative control用の3枚の切片を貼付し、偽(pseudo)反応の有無を厳密に判定する。また複雑な化学反応の繰り返しであるので、再現性を高めるためには、時間と温度を可能な限り厳密に制御しなくてはならない。実験台には温度計を常置し、実験毎に実験ノートに室温を記録する癖をつける。
(2-1-A)固定:屠殺後直ちに臓器を摘出し、固定液中でできるだけ小さく細切する。細切した組織標本を固定液中に浸漬し、1週間固定する。
■チップ:発砲スチロール製の平板上にあらかじめ固定液をたらしておき、そこに摘出した臓器を漬けながら両刃のカミソリで一辺1〜2ミリの立方体になるように切る。両刃のカミソリはジレット社かシック社製のステンレス刃が高価であるが優れている。カミソリ刃は錆止めの鉱物油が塗布されているので、使用前に清浄アセトンで洗浄して、丁寧に鉱物油を取り除いておくかないと、鉱物油が組織標本に付着してしまう。意外に思われるが、これは重要なポイントである。
■チップ:固定液としては、10%中性緩衝ホルマリン固定液(和光純薬などから市販品が販売されている)、4%パラホルムアルデヒド固定液、Omni-Fix固定液(American
Histology Reagent社:高価であるが優れている)などで浸漬固定すれば標的のmRNAやDNA等の核酸と組織構造の保存が良い。ただし、酢酸やピクリン酸などの酸性の固定剤(古いホルマリン液にはホルムアルデヒドが酸化して生成した蟻酸が含まれることに注意)を含むBouin液などの固定液、水銀などの重金属を含むZenker液などの固定液はRNA・DNAが非特異的に分断されてしまうので不適切である。アセトン、エタノールなどの有機溶媒系固定液は、核酸の保存がよいので細胞標本に適しているが、組織構造の保存に難点があるので細胞を観察する場合には採用を考慮するとよいが、組織標本の固定には不適切である。
(2-1-B)包埋と薄切:常法に従って組織包埋専用のパラフィン(Merk社:高価であるが優れている)に包埋し、厚さ3μm以下の薄切片を作成し、シランコートスライドグラスに貼付する。
■チップ(シランコートスライドグラスの自作法):シランコートスライドグラスはマツナミ社などから市販されているが、組織切片が剥がれやすい場合には、使用直前に自作するほうが成績がよい。自作は簡単である。汎用の病理組織検査用スライドグラス(マツナミ社)をchromerge洗剤(American
Histology Reagent社)などのアルカリ性洗剤に一昼夜浸漬後、60℃30分間ソニケーターで2回洗浄し、さらに1時間流水洗浄後ミリQ水にて充分に洗浄し、180℃2時間乾燥する。室温まで冷却後2%(v/v)シラン(3-aminopropyl-triethoxysilane;
Sigma Aldrich社)-アセトン溶液(液体クロマトグラフ用超精製級)に室温10分間浸漬する。アセトンで2回、ミリQ水で1回軽くリンスしてから乾燥器で60℃一昼夜乾燥し、シリカゲルを入れた密閉容器中に使用時まで保存する。シランコートしたスライドグラス表面にはゴミが付着し易くなっている。一度付着してしまうと剥がれない。スライドグラスの保存には細心の注意をはらうこと。
■チップ:PCR操作中に切片が剥がれることを防ぐため、切片(厚い切片は剥がれやすい)をシランコートスライドグラスに貼り付けた後、一晩45℃のホットプレート上で加温し、その後60℃30分間ヒートアップする(切片のまわりのパラフィンが溶ける)。その後45℃に冷却し、スライドグラスを3分間キシレンに浸漬してパラフィンを除去する操作をパラフィンが完全に除去するまで繰り返した後(普通は5回繰り返せば充分)、100%EtOHに5分間浸漬した後充分に乾燥器内で乾燥する。
■チップ(100%EtOHの作製法):モレキュラーシーブで完全に脱水した液体クロマトグラフ用超精製級EtOHを作製する。意外に知られていないことだが、EtOHは購入した時点で0.5〜1%ほど水を含んでいる。
なお、液体窒素で急速凍結した臓器標本を用いる場合は、クリオスタットを用いて厚さ5μm以下の薄切片を作製し、シランコートスライドグラスに貼付し、冷風にセットしたヘヤードライヤーを用いて素早くする風乾する。その後、切片を貼付したスライドグラスを氷冷4%パラホルムアルデヒド固定液あるいはOmni-Fix固定液に1時間浸漬して固定した後ミリQ水で充分に洗浄した後プロテアーゼ消化に供する。
(2-1-C)プロテアーゼ消化:固定された組織切片では、検出しようとするDNA/RNAがタンパクに埋没しており、このままではPCR反応などがすすまないので、軽くタンパク分解酵素を用いてタンパクを溶かして標的とする核酸を露出する。組織切片を貼付したスライドグラスを湿箱中(アクリル製の組織切片専用のボックスが市販されているが、プラスチック製の密閉できる箱で代用できる)に水平をたもって置き、パップペン(マツナミ社)で切片を取り囲むようにマークした後、切片を十分に覆う量(約300μl/切片)のpepsin液(2 mg/ml pepsin-0.1 N HCl;和光純薬)を静かに滴下する。湿箱を密閉し、20℃90分間消化する。次いで、0.1% (w/v) DEPC-ミリQ水にスライドグラスを1分間浸漬して消化反応を停止させる。その後100%EtOHに5分間浸漬した後乾燥し、続いて下記のPCR反応を行う。
■チップ:Pepsinによる消化時間の決定は、in situ PCRの成否を決定する重要なポイントである。酵素活性はロット差がある。組織の病態や固定条件の差異などの諸条件に対応して反応時間を調整しなくてはならない。私たちの経験では、10%中性緩衝ホルマリンで室温1週間固定した多くの臓器の場合、20℃90分間消化するとよい。コラーゲンなどの線維成分の多い標本では消化時間をやや長めに設定する。
■チップ:Pepsinの代わりにprotease Kやtrypsinを用いる方法もあるが、pepsinが最も消化の程度を再現性高く制御しやすい。
■チップ:消化が長すぎると組織が崩れてしまい局在が不明瞭になってしまう。短すぎるとDNA/RNAがタンパクに埋没したままで露出していなので、PCR反応がうまく進行しない。Positive
controlにおいても陽性細胞が観察されない場合、組織構造が崩れている場合は消化時間が長すぎ、逆に構造がよく保たれている場合は、消化時間が短すぎる。
mRNAやRNAウイルスなどを検出しようとする場合には、pepsin消化にひき続いてRNase-free DNase消化(37℃6時間)を行ってあらかじめ偽反応の原因となるDNAを分解しておき、その後に逆転写反応を行ってcDNAを合成する。この反応は通常のエッペンドルフチューブで行う反応と同じ条件の化学反応を上記の湿箱に置いた組織切片上で行う。例えば、oligo(dT)
primer を用い、Amersham Pharmacia Biotech社のT primed first-strand kitを使用してfirst
strand cDNAを合成する。
3. In situ PCR染色法
In situ PCRでは、組織切片上でPCR反応を行って検出しようとするDNAを数百万倍まで増幅しておき、その後、それを組織化学的に可視化する。私たちは、in situ PCR専用サーモサイクラー(GeneAmp In Situ PCR System 1000;Perkin-Elmer社)を用いることで比較的安定した結果を得ている(図1)。これを用いる場合は、装置に添付されているマニュアルに従えばよい。本格的にin situ PCRを行う場合は、専用装置の購入をすすめるが、ここでは手技に熟練を必要とするが一般的なPCR用サーマルサイクラー(0.5 ml-PCR用エッペンドルフチューブをアルミ製ヒートブロックの穴に立てて隙間をミネラルオイルで満たしてPCRを行うタイプの装置でないとin situ PCRに流用できない)を用いても行える簡便法を紹介する。
 |
|
図1 |
(3-1)PCRによる標的DNAの増幅
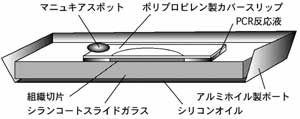
PCR反応液(約3 mm×3 mmの断面の組織切片1枚の染色に約50μl必要)をスライドグラスガラス上に貼付された切片全体に均一となるように滴下し、切片を覆う最小面積の細片としたポリプロピレン製カバースリップで覆い、少量の紅色のマニュキアをスポット状に塗ってカバースリップを保定する(図2)。スライドグラスガラスを自作したアルミホイル製ボート(家庭で料理などのために使用しているごく一般的な薄手のアルミホイルを、スライドグラスを中に包みこむような箱舟状に加工する)入れ、予め80℃に加温してミネラルオイルを滴下したPCR用サーマルサイクラーのアルミ製ヒートブロックの上面に密着するように丁寧に置く。Hot
start型のDNAポリメラーゼを使うことで非特異的反応を軽減できる。その場合の温度サイクルは、例えば94℃4分間上昇後、55℃2分間と94℃1分間のPCRサイクルを20回繰り返した後、スライドグラスを砕氷上に置いて反応を停止させる。
 |
|
図2 |
■チップ:PCRに用いる試薬は、病理組織診断用として市販されているin situ hybridization用キット(例えばGeneAmp;Perkin-Elmer社)に含まれているものを利用すると便利かつ経済的でよい。
■チップ:スライドグラスの底面をアルミホイル製ボートに密着させないと温度制御が精密に行えないのでシリコンオイルを極薄く塗って密着させる。シリコンオイルは熱伝導性が高い。コンピューターのCPUの冷却性能向上のためにCPUチップと金属製放熱フィンの隙間にシリコンオイルを塗るのと同様である。
■チップ:In situ PCRの場合、PCRサイクルは少ないほど組織切片の損傷が軽くかつ特異性が高いので、PCR産物の検出が可能な限り最小回数に止めること。
GeneAmp In Situ PCR System 1000を使用する場合も概ね同様である。すなわち、専用のスライドグラスに組織切片を貼付して上記の消化処理などを済ませたあと、専用のカバーディスク(AmpliCover
disk and clip;Perkin-Elmer社)をクリップで固定し、サーマルサイクラーにセットする。95℃1分処理後、56℃2分-72℃1.5分-95℃1分のサイクルを20〜30回繰り返し、最後に4℃に下げて反応を停止させる。
反応後、カバースリップを丁寧に取り除き、スライドグラスをペーパータオル上に垂直に立ててミネラルオイルを落とした後、清浄な脱水キシレン(EtOHの脱水と同様にモレキュラーシーブを用いて行う)に2分間宛3回浸漬してミネラルオイルを完全に取り除いた後、100%EtOHに5分間浸漬後、乾燥する。
ここで用いたPCR反応液(GeneAmp In Situ PCR System 1000を用いた場合も同様である)の組成は次のようなものである。すなわち、1,000μlの反応液中に100μlのPCR緩衝液、200μlの25
mM MgCl2(MgCl2濃度は増幅しようとするDNAのCG含有量などによって異なる。一般に最終濃度4.5〜5.0 mMである)、160μlのdNTP[各ヌクレオチドとも最終濃度を200μMに調製する。最近、PCR反応液に最終濃度2.5μMのDIG-labeled
dUTP(Roche Diagnostics社)を添加してPCRで増幅されるDNAに部分的にDIGを組み込んで標識する手法が繁用されるようになってきている。ここではDIG-labeled
dUTPを添加する方法を記述している]、40μlの2% (w/v)ウシ血清アルブミン(BSA;Sigma社のDNase freeのものを供すること)、20μlの1
mM digoxigenin-dUTP、40μlの20μM primer-1(forward primer)、40μlの20μM primer-2(reverse
primer)、380μlのミリQ水、20μlのTaq DNA polymerase(最終濃度300 unit/ml)を含む。
■チップ:In situ PCR反応液は、溶液系PCR反応液よりMgCl2、taq DNA polymeraseおよびBSAの濃度が高い。PCR反応がうまく進まない場合、最初にMgCl2の至適濃度を検討するとよい。
■チップ:各プライマーのサイズは20塩基前後が望ましい。
■チップ:Primer-1とprimer-2を除いたものを毎回negative controlとして準備し、非特異的陽性反応の有無を確認しなくてはならない。Positive
controlとしては、一般に全ての細胞が陽性となるハウスキーピング遺伝子のprimerセットを設計して添加する。別途スライドグラスを用意し、プロテアーゼ消化に続いてPCR反応を行う前にRNase-free
DNaseにてDNA消化を行って、PCR反応が陰性となることを確認しておく。
■チップ:PCRのサーマルサイクルは、溶液PCRと同様に温度設定し、各温度での反応時間を約30秒ほど長めに設定して行うと成績がよい場合が多い。サイクルは20〜30の間で最適サイクルを決定するとよい。サイクルが30回を増えると組織の痛みが激しくなり、かつ非特異的反応が増える。ただし、in
situ PCR専用サーモサイクラーを用いる場合は、その説明書に従うとよい。
(3-2)標識DNAの可視化(免疫組織化学的染色)
PCR産物に対してhybridization反応を行うことが本来のin situ PCR-hybridization染色であるが、上述のようにPCR反応中にPCR産物に直接DIG標識することで、hybridization反応を省略できる。この場合、PCR産物の結果を可視化することは、ごく一般的な免疫組織化学染色と同様であり、容易である。
PCR反応後、切片を0.2% (w/v) BSAを含む1×SSC溶液で52℃5分間リンスした後、ブロック溶液[5% (w/v) スキムミルク、5%
(w/v) BSA、20μg/mlマウスIgGおよび20 mg/mlの組織切片を作製した動物種のIgG(全血清でも可能)を含むPBS(pH 7.4)]を約200μl/切片ずつ静かに滴下し、湿箱内にて室温30分間反応させる。反応後キムタオル上にスライドグラスを垂直に立てて余分な溶液を捨て(決して切片を乾燥させてはならない)、洗浄せずに急ぎ切片上にブロック溶液にて400倍に希釈した西洋ワサビペルオキシダーゼ標識マウスモノクロナール抗DIG抗体(Roche
Diagnostics社)を約100μl/切片ずつ静かに滴下する。湿箱内で4℃18時間反応後、0.02% Tween20-PBS(pH 7.2)、続いて0.1
M Tris-HCl緩衝液(pH 7.2)にて充分にリンス(3〜4回で充分)し、ペルオキシダーゼ反応発色液(0.05% (w/v) diaminobenzidine-0.02%
(v/v) H2O2-100 mM Tris-HCl, pH 7.2)を滴下する。滴下後直ちにスライドグラスを顕微鏡のステージにセットし、1枚毎丁寧に観察しながら非特異的反応が出ない適切な時点で反応を停止する(毎回温度と時間を記録しておき、再現性を高める)。スライドグラスを蒸留水で1分間リンスして反応を停止させる。
0.5% (w/v) メチルグリーン-100 mM酢酸ナトリウム-酢酸緩衝液(pH 4.0)にて淡く核のみが染まるように(室温10分間ほどでよい)対比染色(カウンター染色とも呼ばれる)し、蒸留水にて分別する。100%ブタノール(MeOHやEtOHで脱水すると脱色されてしまう)にて脱水後、キシレンに透徹し、エンテラン(Merk社)にて封入し、検鏡する。
■チップ:抗体を標識する酵素は、アルカリホスファターゼ、グルコースオキシダーゼなどの植物由来で安定な酵素ならいずれの酵素でも使用可能である。ただし、西洋ワサビペルオキシダーゼに代わってアルカリフォスファターゼにて標識したマウスモノクロナール抗DIG抗体を用い、nitroblue
tetrazolium/5-bromo-4-chloro-3-indolyl phosphateを基質として発色させる方法は、特異性に優れているが、キシレン透徹後エンテラン封入することが困難なためグリセリン封入しなくてはならない。操作ステップが多いin
situ PCR後の組織切片はかなり痛んでいることが多い(特に、胎児の組織は傷みやすい)ので、グリセリン封入をした場合には標本の保存性が悪く、観察が困難になることが多い。初めてin
situ PCRを行う場合には、PCR産物の可視的検出にはRoche Diagnostics社からDIG nucleic acid detection
kitとして市販されているものを供することを推奨するが、これはin situ hybridization用に開発されたもので、アルカリフォスファターゼ標識抗体を用いるため組織の保存が悪い。
■チップ:抗体の反応を37℃1時間や室温1時間という方法もある。これは特異性を犠牲にしても迅速性が重要な病院での病理診断用に開発された方法である。実験室レベルでは、特異性の高い低温で長時間反応させる方法を推奨する。
■チップ:最近では、優れた蛍光色素が多く開発されてきており、発色反応のステップが省略できる蛍光色素にて標識したマウスモノクロナール抗DIG抗体を用いる手法が広まってきている。
■チップ:In situ PCR後の組織切片がかなり痛んでいる場合には、操作ステップを少なくするために対比染色を省略するとよい。PCR産物が少ないために発色が薄い場合も省略するとよい。対比染色を省略した場合、微分干渉コンデンサーが装着された正立型光学顕微鏡で組織切片を観察すれば、組織構築の観察になんら支障はない。
4.おわりに
In situ PCR法は、従来からの方法では困難であったごく僅かに存在するDNA/RNAを細胞単位で可視化して調べるのに有効である。本法は検出感度が極めて高いので、将来様々な分野で応用されるものと考えられるが、未だ安定した反応を得るためには熟練を要する開発途上の特殊技術である。最近、GeneAmpなどの専用試薬キットやin situ PCR専用のサーモサイクラーなどの専用器具の開発が進んでおり、遠からず一般のラボでも容易に実施できる技術として普及するものと期待している。
参考文献
1) Nuovo GJ. PCR in situ hybridization: Protocols and
applications 3rd ed, Lippincott-Raven Press, New York, 1997.
2) Bagasra O, Hansen J. In situ PCR techniques, Wiley-Liss, New York,
1997.
3) Nuovo GJ. PCR primer: A laboratory manual, Cold Spring Harbor Lab, Plainview,
1995.
4) Nuovo GJ, Becker J. PCR application in pathology, Oxford Univ Press, Oxford,
1995.
5) Nuovo GJ, Frontiers in Bioscience 1996. 1: 4.
6) 眞鍋昇ら.実験医学 1997. 15: 103.
7) Manabe N et al. Hepatology 1996. 24: 366.
8) Manabe N et al. Hepatology 1995. 22: 267.
9) Manabe N et al. Hepatology 1993. 18: 1344.